I started writing a post earlier today about some cool collaborative genetics projects that are going on around the world, and then I realized that these projects all rely on a common knowledge of sequencing. In an effort to not completely sound like a wikipedia article on sequencing, I’m going to just focus on the ones that I use in the lab on a fairly regular basis, and Next-gen sequencing is sort of like, “you seen one you seen ‘em all” type of technology. So I’m going to go through in chronological order, Sanger (or Shotgun sequencing), Next Generation/High Throughput Sequencing (with the emphasis being on Illumina’s method), and then touch briefly on Next-next-gen sequencing (I think they’re calling it third generation now.) Then I’ll take a brief look at how we use sequencing in the lab. Ironically now, we all use High Throughput Sequencing as our first pass through, but everything called in HTS, and I mean everything will get verified through Sanger sequencing, the oldie but goodie.
Shotgun sequencing
Shotgun sequencing
Shotgun sequencing, also known as Sanger sequencing because it was developed by some dude named Sanger. Here’s how it works. You amplify the sequence you want, whether by PCR or a vector in vivo, and then your sequencing reaction contains your purified target sequence, primers for that sequence, DNA polymerase, regulary ol’ dNTPs, and then flourescently labeled dNTPs, which also happen to be chain terminating, which means after one is added, no other dNTPs will be added on after that. Your sequence will stop after a flourescently labeled dNTP is added. See where I’m going with this yet? So you put all that stuff in a tube and start a PCR reaction cycling (I know, I know, polymerase chain reaction reaction, but it’s like ATM machine, you just sometimes need to say the machine after it even though it’s included in the acronym), and what happens is, polymerase works its magic, attaching dNTPs and labeled dNTPs on to the exposed single strand of your target DNA sequence. Except for that, every time a labeled dNTP is added on, that DNA polymerase stops putting on more dNTPs, so it’s a set length. So after x amount of cycles, what happens is you’ll have a whole bunch of fragments of your sequence, all with one labeled nucleotide as the last one on that sequence, like thus:
Your labeled sequences are then sorted by size, electrophoresis, in a capillary tube, and the colors are read off one by one, and the data you get back looks like this:
So there you have it. Sanger sequencing. Let’s recap.
Pros: For small sequences? It’s cheap and fast. I can throw my product in a microcentrifuge tube with some primers, walk it down to our sequencing facility, and for 8 bucks, they’ll give me really high quality sequence of about 800 base pairs or so.
Cons: You need to know your target sequence (or at least, enough to amplify, and sequence) which means you need to already know the genomic location, and you need to design primers flanking.
Where it’s been: People got whole genome data from these little dudes. That’s where the shotgunning comes in. You get massive amounts of these short fragments, and then some crazy algorithm + a super computer + someone smarter than me will sit there and align sequence fragments by where they overlap, and construct a whole genome sequence from that. No one does that.
Where it is now: People use Sanger sequencing for much smaller applications now. We use it to verify PCR fragments, verifying vector sequences. We’ll also use it to verify some of our sequence calls that we get from the high through put methods of DNA sequencing.
Next Generation Sequencing (NGS)
Next Generation Sequencing has a couple of aliases. Some people call it High Throughput Sequencing, others call it, well, I spoke too soon. I think people just call it next gen or high throughput. There are a bunch of platforms for it, which is great. Next gen sequencing is the textbook definition of how capitalism works. Competition to come up with the best product for the lowest price is allowing for the development of some genius technologies at prices that are allowing more and more people to use this an integral part of their research. That being said, I’m really only going to talk about Illumina’s platform, because it’s what my lab uses, and it’s the one I’m most familiar with. The actual technologies are a bit different, but the concept of high throughput/efficiency, is the same for all.
There are two big steps in NGS, I like to think of it as “at the bench” and then “at the really scary expensive sequencing machine”, but I’m not requiring that you use my terminology. At the bench is preparing the genomic library, and at the really scary expensive sequencing machine is cluster generation and the actual sequencing.
At the bench
Preparing genomic library: You start out with your genomic DNA, you shear it into more manageable sizes (~300 bp), and you ligate Illumina’s sequencing adapters on to them. This is your first step, and these sequencing adapters will be necessary for the sequencing steps. You then amplify these sequences. The sequence that you use to amplify are the same as the sequence adapters that you ligated on, which ensures that your fragments have both of their ends ligated.
Illumina Sequencing
The Machine: Your prepared genomic library is placed on a flow cell. A flow cell has eight lanes, and in general, one sample is run per one lane.
Cluster Generation: In this part, your single-stranded fragments randomly attach to the inside the flow cell channels, remember those adapters you ligated on to prep your library? Those primers are on the surface of that flow cell.

Unlabeled nucleotides are then added, as well as an enzyme that initiates solid-phase bridge amplification. This just means those free standing ends bend over to find their other primer, like this:
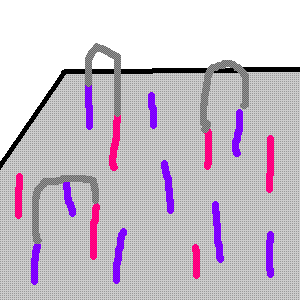
The enzyme also works to then make all of your little single bridges into double stranded bridges. The double stranded molecules are then denatured, leaving only single stranded templates attached to the flow cell (but because you made them double stranded before denaturing, you have complementary strands.) You repeat this about a million times, so you end up with several million dense clusters of double-stranded DNA.
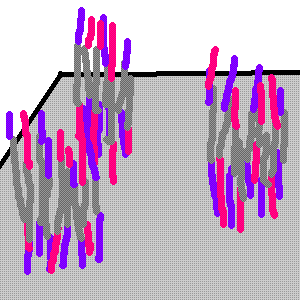
You end up choosing only one sequence adapter, so you result in a cluster of only direction.
The Actual Sequencing
Primers are added plus all four labeled dNTPs, which again, are made so that only one base can be added per cycle. When the first primers, dNTPs, and DNA polymerase is added to the flow cell, a laser is used to excite the fluorescence, and an image is captured of the emitted fluorescence, the images look something like this:
Then you literally, rinse, and repeat. Rinse to remove all the leftover dNTPs that didn’t stick on that last time, oh but then you have to remove the terminator property of the dNTPs that are stuck on to allow further extension, add more dNTPs, and take another picture.

See? Same place (those are your clusters) but a new color, and a new picture is taken.
Cycles continue to give you 76 reads. Computers analyze the image data to give you actual sequence. The cool thing is is that they use astronomy imaging techniques to monitor the same place over time. After you analyze all your millions of clusters to get your reads, you have real whole genome sequence!
Again, this is only just sequence. You don’t know where in the genome it aligns to, only that it existed in your original sequence. People use a variety of programs to align sequences to a reference genome. I’ll go a little into aligning after I briefly touch on a variation on this theme.
Exome sequencing: A lot of times, groups will choose just to sequence the “exome”, that is, all of a person’s exons. The bet is, that this is where the good stuff is going to be, mutations in coding regions are definitely A Very Bad Thing, and also way easier to functionally analyze and decide whether or not it’s a causative bad change. Plus, it’s easy. You add a sequence capture step while you’re preparing your library, so you only capture the exons.
Why exome and not whole genome? The main reason is $$$$$. Exons make up about 50 mb of the genome, as opposed the 3 gb. (mb=megabase, gb=gigabase: that’s 50 million basepairs compared with 3 billion basepairs) It’s about $3k to sequence all the exons of a person with Illumina, about ~$10k to sequence everything. Or something like that. The prices are going down each day though. Plus, the most informative information is coming from your coding regions anyway. A lot of times, when people do whole genome sequencing, they filter out all the noncoding regions anyway. It’s a lot of data, and it’s easier to tackle parts that you know are important. If you can’t find anything then, it’s time to look in noncoding regions. But we know so little about the genome, more information is not always better.
Pros: Next gen sequencing is the crux of "discovery" science, that is, looking for something in the genome that you have no idea where it is, or even if it's there. It generates massive amounts of data which brings me to...
Cons: The shear amount of data that you get from this means that it takes a huge team, huge computer power, to sort through all the data, and there are still large amounts of unaligned sequence that no one can make head nor tail out of. And for all you know, the piece that you're looking for is in that little nugget on your hard drive that you don't know what to do with. And it's expensive.
Caveats
The reference sequence: The “one” reference sequence that people use---in general, as used by the UCSC browser, is this one unknown guy in like, Buffalo, NY. See any issues with that??? There are an insane amount of issues with that. When we use this reference, we are taking a huge huge chance that this is just the normal framework of the human genome. This is really most likely not the case. There are now a ton of databases that are purely dedicated to documented all variation that has been reported in the human genome. dbSNP, 1000 genomes, hapmap, these are huge collaborative projects with the sole purpose to work with and around the fact that people are so different, there’s no way we can have just one reference sequence to align to.
Speaking of aligning, there are two words that people use to describe what they do with their short little sequences from the sequencing machine. They usually say that the next step is aligning or assembling the reference. A lot of the time people use them interchangeably, so much so that it’s pretty much accepted, but it’s not, there’s a subtle difference. Aligning your sequence to a reference implies just that: that based on your sequence, you look at the reference sequence, and see where that piece goes, and you put it there. When you assemble your own sequence, you’re putting the pieces of your sequence together based on the parts of those sequences that overlap each other. That’s how the first “shotgun” sequences were created together.


That concludes this super brief (well, I tried to make it super brief, I’ll give me...a C for effort) background on sequencing. Next, I’ll look at what people are doing with these technologies. Stay tuned for stuff on the 1000 Genomes Project and HapMap.
References: I got most of this from my head, and some powerpoint slides left over from classes I took in college. Unfortunately, my prof cited a post doc, who didn’t cite anything so the google images that I’ve been basing my paint images off of were found here
No comments:
Post a Comment